














Almira Bartolomé
Directora de Business Development, 3PBIOVIAN

Jurgen Van Broeck
CEO, mAbxience

Cristina López
Directora de la oficina de Barcelona y Chief Research & Development Officer, Qualipharma



Abordaje del rol del promotor en los estudios clínicos observacionales con medicamentos de uso humano
Los estudios observacionales con medicamentos de uso humano son una herramienta clave para la obtención de datos sobre sus condiciones de uso, seguridad y efectividad en el contexto de práctica clínica real. Estos estudios tratan de aportar información complementaria a la existente acerca de los beneficios, riesgos o el uso terapéutico de los medicamentos a estudio.
El texto refundido de la Ley de garantías y uso racional de los medicamentos y productos sanitarios, aprobado por Real Decreto Legislativo 1/2015, de 24 de julio, diferencia un estudio observacional y un ensayo clínico, en función de la asignación de la estrategia terapéutica. En un estudio observacional, los medicamentos administrados a los participantes no son definidos por el protocolo del estudio – como ocurriría en un ensayo clínico - sino por la práctica habitual, de acuerdo con las condiciones establecidas en su autorización. Asimismo, tal y como se describe en el Real Decreto 577/2013, de 26 de julio, estas condiciones deben ser respetadas durante todo el periodo de realización en aquellos estudios de seguimiento prospectivo.
En los últimos años, la evolución de la normativa europea y española recoge los cambios en los requisitos y procedimientos requeridos para la realización de los estudios observacionales con medicamentos. A nivel europeo, destacan las Directrices sobre Buenas Prácticas de Farmacovigilancia europeas, en relación con los estudios posautorización de seguridad de tipo observacional, en las que se recogen las obligaciones de los titulares de autorización de comercialización de estos estudios.
En el ámbito estatal, el término de estudio observacional es introducido por el Real Decreto 1090/2015, de 4 de diciembre, por el que se regulan los ensayos clínicos con medicamentos, los Comités de Ética de la Investigación con medicamentos y el Registro Español de Estudios Clínicos, con el objetivo de adecuarse a lo descrito en el Reglamento (UE) número 536/2014 del Parlamento Europeo y del Consejo, de 16 de abril de 2014.
Asimismo, nace la necesidad de agilizar los procedimientos y trámites administrativos relacionados con los estudios observacionales con medicamentos, basados en la práctica clínica habitual. En el ámbito estatal, el Real Decreto 957/2020, de 3 de noviembre, por el que se regulan los estudios observacionales con medicamentos de uso humano, recoge los requisitos y procedimientos necesarios para su realización.
Con base en la normativa vigente en España para la realización de estudios observacionales con medicamentos, el citado Real Decreto 957/2020, se define este tipo de estudios como toda investigación que no cumpla las condiciones de ensayo clínico, siendo las mismas: determinar los efectos beneficiosos de los medicamentos; identificar, caracterizar, cuantificar las reacciones adversas y medir su efectividad; y obtener información sobre patrones de utilización de los medicamentos en la población. Asimismo, una de las características diferenciales clave de este tipo de estudios es su realización acorde a las condiciones de práctica clínica habitual. Además, en el presente Real Decreto se incluye la definición de estudio observacional con medicamentos de seguimiento prospectivo, los cuales implican el seguimiento de los participantes hasta la obtención de la variable resultado, que no se ha producido en el inicio del estudio.
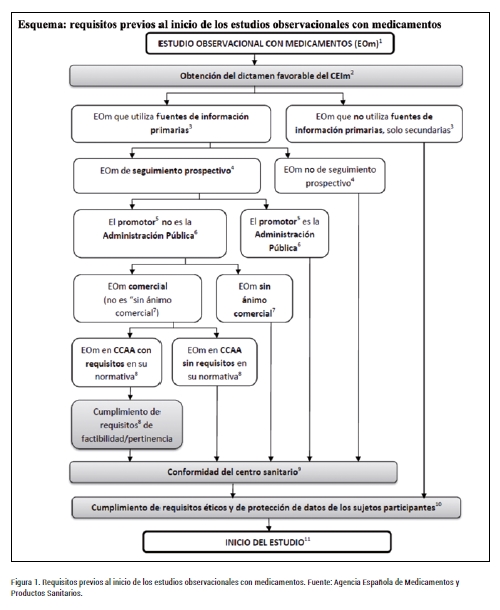
El Real Decreto 957/2020 introduce también modificaciones en los requisitos previos a la realización de este tipo de estudios (Figura 1), como la anulación de la autorización previa de la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS); o la eliminación de la clasificación de los protocolos de los estudios. Los requisitos exigidos incluyen la obtención del dictamen favorable de un CEIm acreditado en España, la conformidad del centro o centros sanitarios participantes, y el cumplimiento de requisitos éticos y de protección de datos de los sujetos. Además, aquellos estudios de seguimiento prospectivo podrían someterse a los requisitos adicionales establecidos por las comunidades autónomas de los centros participantes.
Acorde a lo descrito en la normativa vigente, los responsables del estudio son el promotor y el investigador principal, o los investigadores colaboradores si se trata de un estudio multicéntrico. En caso de ser preciso, pueden contar con la participación de un equipo colaborador no investigador.
Concretamente, el promotor se define como cualquier “individuo, empresa, institución u organización responsable de iniciar, gestionar y organizar la financiación de un estudio observacional con medicamentos”. En el desarrollo de sus estudios, deberán tener en cuenta las directrices de la Comisión Europea y las instrucciones para la realización de estudios observacionales con medicamentos que publique la AEMPS en su página web, y que elaborará en colaboración con los CEIm y las administraciones sanitarias competentes de las comunidades autónomas.
De este modo, el artículo 9 del vigente Real Decreto 957/2020 recoge las principales obligaciones del promotor. En primer lugar, deberá identificar las fuentes de financiación del estudio, así como la disposición de los medios necesarios para su realización. Deberá firmar, modificar y facilitar el protocolo al equipo investigador. Asimismo, deberá realizar las solicitudes necesarias para cumplir con los requisitos previos a la fecha de inicio del estudio de acuerdo con los procedimientos aplicables. El promotor también deberá realizar comunicaciones con el CEIm que emitió el dictamen favorable y las autoridades sanitarias pertinentes, presentando el informe de situación, el informe final de resultados, así como la notificación de una interrupción temprana del estudio.
Abordando las obligaciones relacionadas con los datos del estudio, el promotor deberá asegurar la fiabilidad de los mismos mediante la realización de los controles de calidad pertinentes. Deberá garantizar la publicación de aquellos estudios de seguimiento prospectivo en el Registro Español de Estudios Clínicos. Además, deberá publicar los resultados, así como informar de los resultados que modifiquen la relación riesgo-beneficio de los participantes.
Asimismo, en todo caso deberá respetar la confidencialidad de los datos de los sujetos, así como comunicar las sospechas de reacciones adversas a las autoridades sanitarias, de acuerdo con lo establecido en artículo 15 del presente Real Decreto. Finalmente, el promotor deberá conservar el contenido de los datos del estudio en el archivo maestro, y facilitar posibles inspecciones de autoridades sanitarias.
Por todo lo anteriormente expuesto, la implicación de la figura del promotor en la realización de este tipo de estudios requiere un elevado conocimiento de la normativa vigente, tanto estatal como europea, así como la participación coordinada de un equipo multidisciplinar, con el objetivo final de aportar nuevos conocimientos acerca de medicamentos de uso humano presentes actualmente en la práctica clínica habitual.
Bibliografía y normativa:
- Instrucciones sobre procedimientos, plazos y formato de los datos que incluirá? el Registro Español de estudios clínicos (REec) para la publicación de información sobre los estudios observacionales con medicamentos. Versión 14 abril 2021.
- Memorando de colaboración entre los Comités de Ética de la investigación con medicamentos para la evaluación y gestión de los Estudios Observacionales con Medicamentos. Versión 1 noviembre de 2021.
- Preguntas y respuestas sobre el real decreto 957/2020, de 3 de noviembre, por el que se regulan los estudios observacionales con medicamentos de uso humano. Versión 3 – 3 de marzo de 2021.
- Real Decreto Legislativo 1/2015, de 24 de julio, por el que se aprueba el texto refundido de la Ley de garantías y uso racional de los medicamentos y productos sanitarios.
- Real Decreto 1090/2015, de 4 de diciembre, por el que se regulan los ensayos clínicos con medicamentos, los Comités de Ética de la Investigación con medicamentos y el Registro Español de Estudios Clínicos.
- Real Decreto 577/2013, de 26 de julio, por el que se regula la farmacovigilancia de medicamentos de uso humano.
- Real Decreto 957/2020, de 3 de noviembre, por el que se regulan los estudios observacionales con medicamentos de uso humano.
- Reglamento (UE) número 536/2014 del Parlamento Europeo y del Consejo, de 16 de abril de 2014.
| Nombre | Marta Valverde |
|---|---|
| Empresa | Konexio Biotech. |
| Cargo | Responsable de operaciones clínicas |

















Si continúas navegando, aceptas su uso.
Más información





Política de privacidad | Cookies | Aviso legal | Información adicional| miembros de CEDRO