














Aniol Gurrera
CEO, RAYPA

Almira Bartolomé
Directora de Business Development, 3PBIOVIAN

Jurgen Van Broeck
CEO, mAbxience



Así se Diseñan los Nuevos Materiales y Bioterapéuticos: El Poder de la Simulación Computacional
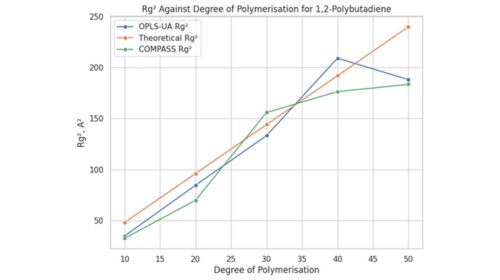
En el contexto del modelado y simulación computacional, diversas metodologías han sido desarrolladas para abordar problemas en el diseño de materiales y bioterapéuticos. Las herramientas computacionales avanzadas permiten explorar propiedades moleculares, optimizar diseños y mejorar la eficiencia de nuevos materiales y fármacos. A continuación, se presentan tres enfoques fundamentales: la modelización de polímeros de precisión, la simulación de la solvatación molecular mediante COSMO-RS y el diseño de bioterapéuticos con inteligencia artificial.
Modelado de Polímeros de Precisión
El diseño y optimización de polímeros involucra la simulación de su comportamiento a nivel molecular para predecir propiedades macroscópicas relevantes. Con la evolución de herramientas computacionales como BIOVIA Materials Studio, los investigadores pueden modelar estructuras poliméricas con alta precisión.
Uno de los enfoques principales en este ámbito es la aplicación de campos de fuerza avanzados, como COMPASS y OPLS-UA, que permiten describir interacciones intra e intermoleculares con mayor fidelidad. La dinámica molecular (MD) y los métodos de Monte Carlo (MC) son empleados para analizar propiedades clave como la densidad, el radio de giro y la temperatura de transición vítrea (Tg), lo que resulta fundamental en la selección de materiales para aplicaciones específicas.
La incorporación de algoritmos de automatización en el modelado de polímeros permite evaluar grandes conjuntos de estructuras, optimizando tiempos de cálculo y garantizando la reproducibilidad de los resultados. Además, la simulación de interfaces poliméricas bajo condiciones extremas, como altas temperaturas y presiones, ha cobrado importancia en el diseño de materiales resistentes y funcionales.
El modelado de copolímeros y mezclas poliméricas es otra área en expansión, ya que permite predecir la compatibilidad entre diferentes componentes y su influencia en las propiedades finales del material. Gracias a herramientas como scripting en BIOVIA Materials Studio, los investigadores pueden diseñar estrategias para optimizar la composición de polímeros de precisión con aplicaciones en tecnología biomédica, electrónica y envases sostenibles.
Simulación de Solvatación con COSMO-RS
El modelo COSMO-RS (Conductor-like Screening Model for Real Solvents) es una metodología que combina la teoría de funcionales de la densidad (DFT) con modelos estadísticos para predecir propiedades termodinámicas de solvatación. Implementado en herramientas como BIOVIA COSMOtherm, este enfoque permite analizar la solubilidad y el comportamiento de moléculas en distintos solventes sin necesidad de experimentación extensiva.
COSMO-RS se basa en el cálculo de la carga superficial polarizada de una molécula en un entorno continuo conductor, seguido de un tratamiento estadístico que permite obtener datos como coeficientes de actividad, particiones entre fases y energías libres de solvatación. Su aplicación es fundamental en el diseño de formulaciones farmacéuticas, permitiendo optimizar la solubilidad de principios activos y mejorar su biodisponibilidad.
El modelo también es clave en la selección de solventes para procesos industriales, facilitando la identificación de alternativas sostenibles en la química verde. En la industria de los materiales, COSMO-RS permite predecir la estabilidad de mezclas y la compatibilidad entre diferentes componentes, contribuyendo al diseño de materiales innovadores con propiedades optimizadas.
Un aspecto destacado de COSMO-RS es su capacidad para analizar interacciones hidrofóbicas y su relevancia en sistemas biológicos y químicos. Esto ha permitido avances en el diseño de membranas para separación de fases, así como en la comprensión del comportamiento de biomoléculas en soluciones acuosas y orgánicas.
Diseño de Bioterapéuticos con Inteligencia Artificial
El diseño de bioterapéuticos ha experimentado un avance significativo con la integración de inteligencia artificial y aprendizaje profundo.
BIOVIA Discovery Studio ha incorporado herramientas avanzadas que permiten modelar interacciones biomoleculares y predecir la estabilidad de bioterapéuticos de manera eficiente.
Metodologías como RFDiffusion y ProteinMPNN han revolucionado el diseño de estructuras proteicas mediante técnicas generativas. RFDiffusion emplea modelos basados en difusión para refinar estructuras proteicas, permitiendo la generación de conformaciones con estabilidad y funcionalidad optimizadas. Por su parte, ProteinMPNN utiliza redes neuronales para predecir secuencias de aminoácidos con propiedades específicas, facilitando el desarrollo de nuevas terapias basadas en proteínas.
Estas herramientas permiten predecir la estabilidad proteica, evaluar interacciones con biomoléculas y optimizar propiedades farmacocinéticas. La combinación de simulaciones computacionales con inteligencia artificial ha demostrado su eficacia en la aceleración del descubrimiento de fármacos y en la optimización de diseños para una mejor biodisponibilidad y menor inmunogenicidad.
Un campo de aplicación relevante es el desarrollo de anticuerpos terapéuticos, donde la IA permite analizar y mejorar la afinidad de un anticuerpo por su diana biológica. Gracias a la simulación molecular, es posible realizar modificaciones estructurales que optimicen la estabilidad y eficacia de estos bioterapéuticos antes de pasar a pruebas experimentales.
Conclusiones
El modelado computacional ha permitido avances sustanciales en la ciencia de materiales y la biotecnología, proporcionando herramientas para el diseño racional de polímeros, la predicción de solvatación molecular y la optimización de bioterapéuticos. Herramientas como BIOVIA Materials Studio, COSMOtherm y Discovery Studio han facilitado la aplicación de estos enfoques en diversos sectores industriales y académicos.
A medida que la tecnología avanza, la integración de inteligencia artificial con simulaciones cuánticas y dinámica molecular promete mejorar la predicción de propiedades moleculares y acelerar el descubrimiento de nuevos materiales y fármacos. La combinación de estos enfoques continúa redefiniendo los límites de la investigación computacional en la ciencia de materiales y la bioingeniería
| Nombre | Noelia Pérez Arcos |
|---|---|
| Empresa | Addlink Software Científico |
| Cargo | Chemical Product Manager |
















Si continúas navegando, aceptas su uso.
Más información





Política de privacidad | Cookies | Aviso legal | Información adicional| miembros de CEDRO