














Aniol Gurrera
CEO, RAYPA

Almira Bartolomé
Directora de Business Development, 3PBIOVIAN

Jurgen Van Broeck
CEO, mAbxience



Inteligencia artificial y modelos fundacionales, la nueva era en productos sanitarios: regulación, validación y monitorización
La Inteligencia Artificial (IA) ya forma parte integral de nuestras vidas y no es ciencia ficción. Desde la utilización de asistentes virtuales personales para organizar nuestra jornada laboral, viajar en vehículos autónomos, hasta en el ámbito sanitario, incluyendo la detección y diagnóstico precoz de enfermedades; la identificación de nuevas observaciones o patrones en la fisiología humana; el desarrollo de diagnósticos y tratamientos personalizados, entre otros, con el objetivo de mejorar la experiencia tanto del usuario como del paciente, la IA es una realidad tangible.
En los últimos años, los modelos fundacionales de lenguaje (Large Language Models, LLM), como ChatGPT, han irrumpido con fuerza en el ámbito de la salud. Su capacidad para comprender lenguaje natural, generar respuestas contextualizadas y analizar grandes volúmenes de información los posiciona como herramientas potenciales de apoyo en diagnóstico clínico, generación de informes médicos, priorización de casos o asistencia en decisiones clínicas. Sin embargo, su integración en aplicaciones (APPs) de soporte clínico plantea enormes desafíos regulatorios. La naturaleza de estos modelos, entrenados sobre datos masivos y basados en el aprendizaje continuo, la diferencia de otras tecnologías médicas y exige marcos robustos para garantizar su seguridad, eficacia y calidad (1).
Este artículo analiza los requisitos regulatorios fundamentales para la validación y monitorización del correcto funcionamiento de APPs que incorporan LLM en contextos clínicos, con referencia a marcos legales europeos (Reglamentos (UE) 2017/745-MDR (2), 2017/746-IVDR (3), 2024/1689-IA Act (4), estadounidense (FDA) (5-7) y otras guías internacionales relevantes (8,9).
¿Qué se entiende por modelo fundacional?
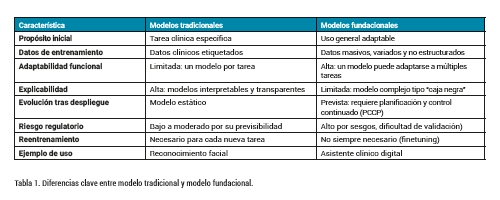
Un modelo fundacional es un tipo de modelo de inteligencia artificial de propósito general entrenado a gran escala con datos masivos y diversos, tanto mediante aprendizaje no supervisado, lo que le permite adquirir representaciones profundas del lenguaje, imágenes, código u otros dominios, lo que da como resultado que el modelo pueda ser posteriormente adaptado a una amplia gama de diferentes usos previstos. Por tanto, y desde un punto de vista regulatorio, un modelo fundacional no puede ser considerado como un producto sanitario ya que tiene un propósito general. Mas bien, debe ser considerado como un elemento que va a utilizarse en el diseño y desarrollo de un producto sanitario con un uso previsto determinado, y que debe ser previamente homologado para asegurar que cumple los requisitos generales de seguridad y funcionamiento relativos al diseño requeridos por el MDR y el IVDR. A continuación, se presenta una tabla comparativa (Tabla 1) que resume las principales diferencias entre los modelos tradicionales de inteligencia artificial y los modelos de IA de nueva generación.
Marco regulatorio aplicable
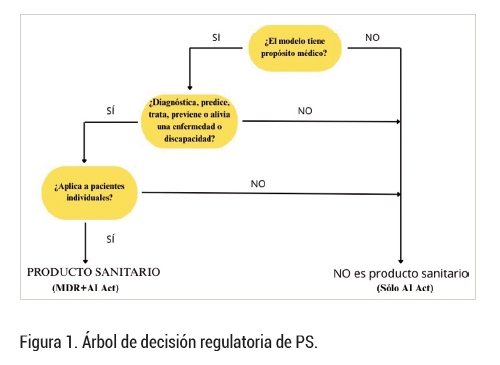
En Europa, los productos sanitarios que incorporan algoritmos basados en inteligencia artificial se regulan bajo el MDR y el IVDR, reglamentos que incluyen dentro de su alcance el producto software con propósito médico específico (SaMD, Software as a Medical Device). El AI Act añade una capa específica de regulación para sistemas de IA de alto riesgo, entre los que se incluyen los productos que deban ser certificados previamente a su puesta en servicio. Este Reglamento aplica también a otras aplicaciones que incorporan inteligencia artificial que podemos encontrar en el entorno sanitario pero que no cumplen la definición de producto sanitario. En la figura 1, se muestra un árbol de decisión útil para decidir el marco regulatorio que aplica en cada caso.
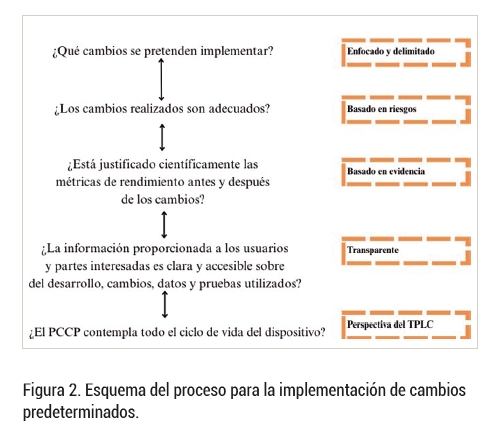
Por su parte, en Estados Unidos, la FDA ha desarrollado un marco específico para SaMD basados en aprendizaje automático, que introduce el concepto de Planes de Control de Cambios Predeterminado (PCCP) (6), que permiten planificar y gestionar cambios adaptativos sin tener que reiniciar todo el proceso de aprobación tras cada actualización. Europa ha adoptado recientemente este enfoque (10), permitiendo que los cambios predeterminados sean evaluados previamente a evaluación de la conformidad. La figura 2, muestra el proceso que se debe seguir para implementar estos cambios:
Sistemática de validación
La validación técnica implica la aplicación sistemática de métodos de evaluación de la calidad de los datos y del modelo para detectar posibles modos de fallo en su comportamiento. Esto incluye métricas orientadas al modelo, como el rendimiento predictivo, la robustez o la interpretabilidad. Asimismo, se integran métricas orientadas a los datos relacionadas con la determinación del tamaño muestral, la dispersión, o el sesgo. Un análisis estadístico riguroso de estas métricas constituye un punto crítico en investigación y desarrollo industrial, y es por ello una pieza clave dentro del proceso de validación técnica.
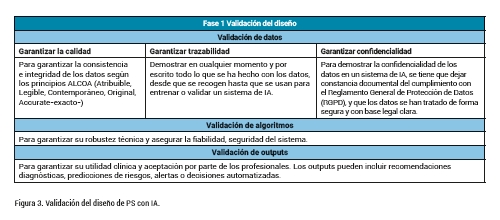
La validación analítica abarca la validación de datos, algoritmos y outputs. La Figura 3 recoge el objeto/alcance de cada fase del proceso de validación.
La validación clínica (figura 4) forma parte esencial del proceso de evaluación de un MDAI, y tiene lugar una vez que el sistema ha sido desplegado en su entorno previsto, ya sea real o simulado. Esta etapa comprende un procedimiento continuo para recopilar, valorar y analizar datos clínicos relacionados con el dispositivo, con el fin de determinar si existe evidencia clínica suficiente para confirmar el cumplimiento de los requisitos esenciales de seguridad y funcionamiento relativos a diseño y desarrollo recogidos en los reglamentos MDR y IVDR. Esta validación requiere no solo la evaluación estadística de resultados, sino también la demostración de utilidad clínica y de su relevancia para los flujos asistenciales. Para ello, diversas iniciativas metodológicas han desarrollado guías específicas para el diseño, implementación y evaluación de intervenciones basadas en IA en distintos tipos de estudio: STARD-AI para estudios diagnósticos, CONSORT-AI para ensayos clínicos aleatorizados, y SPIRIT-AI para protocolos de investigación.
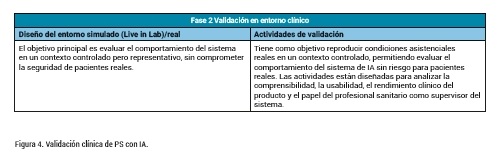
Las principales preocupaciones incluyen si el sistema ha sido validado en una población representativa del contexto clínico de uso (comparabilidad entre datos de entrenamiento y población objetivo), y si las salidas generadas tienen traducción clínica significativa, es decir, si aportan valor diagnóstico, pronóstico o terapéutico útil. En este sentido, resulta clave ir más allá de métricas clásicas como la exactitud, y evaluar aspectos como impacto clínico, aceptabilidad por parte del profesional sanitario, mejora de resultados en salud y aplicabilidad en condiciones del mundo real.
Parámetros para la validación
La validación de un producto sanitario que integra inteligencia artificial, especialmente si se basa en modelos fundacionales y está destinado a decisiones clínicas, debe apoyarse en una batería de parámetros cuantitativos y cualitativos que garanticen su aceptabilidad clínica, técnica y regulatoria. A continuación, se describen indicadores y criterios que se deben considerar.
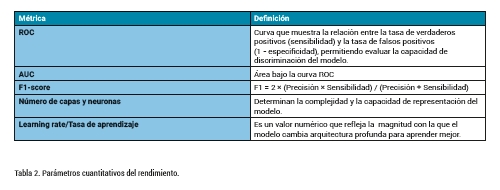
1.Validación del rendimiento analítico: Esta evaluación tiene como objetivo garantizar que el dispositivo cumple con los requisitos de precisión, estabilidad y repetibilidad esperados en su aplicación clínica (Tabla 2).
Se busca responder a cuestiones fundamentales como:
- ¿El dispositivo proporciona resultados fiables y consistentes en diversos escenarios?
- ¿Mantiene su precisión incluso cuando hay cambios en los datos de entrada o en el entorno clínico?
- ¿Sus resultados están alineados con los estándares reconocidos en la práctica médica?
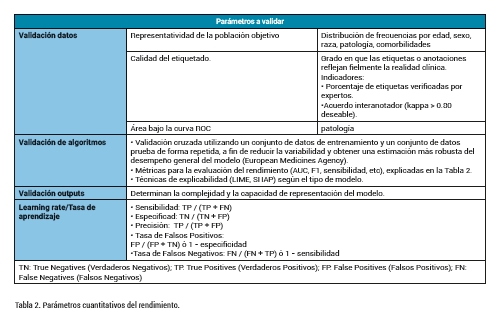
Además de las métricas cuantitativas específicas mostradas en la Tabla 2, es necesario considerar los diferentes niveles de validación implicados en cada componente del sistema. La Tabla 3 detalla estos parámetros, agrupados en tres bloques: validación de datos, validación de algoritmos y validación de outputs, señalando las herramientas y métricas aplicables en cada caso.
2. Evaluación de usabilidad: es recomendable seguir los principios establecidos en la norma ISO 62366, que proporciona un marco para aplicar principios de usabilidad en dispositivos médicos, con el objetivo de identificar, evaluar y reducir riesgos relacionados con el uso. Esta norma ayuda a asegurar que los aspectos de diseño centrados en el usuario minimicen fallos de manejo y favorezcan la seguridad, especialmente en dispositivos que incorporan inteligencia artificial, cuyos resultados pueden influir en decisiones clínicas críticas.
En esta fase se examina la explicabilidad/comprensibilidad del producto. La norma ISO 62366 establece un marco para evaluar si:
- Los usuarios previstos comprenden e interactúan con el dispositivo de acuerdo con su diseño.
- La interfaz y la experiencia de usuario están optimizadas para facilitar su adopción en entornos clínicos.
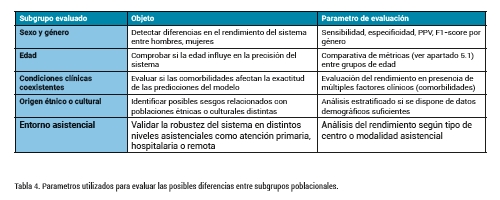
3. Evaluación del rendimiento en subgrupos poblacionales: En el marco de la validación de sistemas de IA aplicados a la salud, resulta fundamental analizar el comportamiento del modelo en distintos subgrupos poblacionales, con el fin de garantizar el cumplimiento de los principios de equidad, no discriminación y robustez clínica. Esta necesidad está recogida en el AI Act, particularmente en los artículos 10 y 15, donde se establece que los sistemas de alto riesgo deben ofrecer garantías de desempeño uniforme en condiciones reales de uso y para todas las personas usuarias previstas. La tabla 4 recoge algunos parámetros habitualmente utilizados en esta evaluación.
El MDR y el AI Act exigen un sistema robusto de vigilancia poscomercialización y un plan de seguimiento clínico poscomercialización. Estos planes deben incluir:
- Monitorización continua del desempeño clínico.
- Detección de errores sistemáticos o efectos indeseados.
- Recolección de datos de uso real.
El despliegue seguro y eficaz de APPs que incorporan LLM depende de cumplir estrictos requisitos regulatorios. Los marcos existentes, MDR en Europa, FDA en EEUU, proporcionan una base sólida, pero es imprescindible adaptarlos a las particularidades de los modelos fundacionales. Gestionar los cambios de forma planificada a través del plan de control de cambios predeterminado y establecer estrategias robustas de monitorización poscomercialización son pasos esenciales.
Referencias
- Bommasani R, Hudson DA, Adeli E, Altman R, Arora S, Arx S von, et al. On the Opportunities and Risks of Foundation Models [Internet]. arXiv; 2022 [citado 14 de julio de 2025]. Disponible en: http://arxiv.org/abs/2108.07258
- Reglamento (UE) 2017/745 del Parlamento Europeo y del Consejo, de 5 de abril de 2017, sobre los productos sanitarios, por el que se modifican la Directiva 2001/83/CE, el Reglamento (CE) n.º 178/2002 y el Reglamento (CE) n.º 1223/2009 y por el que se derogan las Directivas 90/385/CEE y 93/42/CEE del Consejo. Diario Oficial de la Unión Europea, L 117, 5 de mayo,, 2017. p. 1–175.
- Reglamento (UE) 2017/746 del Parlamento Europeo y del Consejo, de 5 de abril de 2017, sobre los productos sanitarios para diagnóstico in vitro y por el que se derogan la Directiva 98/79/CE y la Decisión 2010/227/UE de la Comisión. Diario Oficial de la Unión Europea, L 117, 5 de mayo, 2017. p. 176–332.
- Reglamento (UE) 2024/1689 del Parlamento Europeo y del Consejo, de 13 de junio de 2024, por el que se establecen normas armonizadas en materia de inteligencia artificial y se modifican determinados actos legislativos de la Unión. Diario Oficial de la Unión Europea, 2024/1689, 12 de junio, 2024.
- U.S. Food and Drug Administration (FDA); Health Canada; United Kingdom Medicines and Healthcare products Regulatory Agency (MHRA). Good Machine Learning Practice for Medical Device Development: Guiding Principles. Silver Spring, MD: FDA; October 2021.
- U.S. Food and Drug Administration (FDA). Marketing Submission Recommendations for a Predetermined Change Control Plan for Artificial Intelligence-Enabled Device Software Functions: Guidance for Industry and Food and Drug Administration Staff. Silver Spring, MD: FDA; December 2024.
- U.S. Food and Drug Administration (FDA). Artificial Intelligence-Enabled Device Software Functions: Lifecycle Management and Marketing Submission Recommendations. Draft Guidance for Industry and Food and Drug Administration Staff. Silver Spring, MD: FDA; January 7, 2025.
- Saudi Food and Drug Authority (SFDA). Guidance on Artificial Intelligence (AI) and Machine Learning (ML) Technologies Based Medical Devices. MDS–G010, Version 1.0. Riyadh: SFDA; November 29 , 2022
- Health Sciences Authority (HSA). Software as a Medical Device (SaMD). A Life Cycle Approach. Singapore: HSA; 2022.
- Medical Device Coordination Group (MDCG). MDCG 2025-6: Interplay between the Medical Devices Regulation (MDR), the In Vitro Diagnostic Medical Devices Regulation (IVDR) and the Artificial Intelligence Act (AIA). Brussels: European Commission; june 2025.
| Nombre | Ilham Ben Yahya y Miguel A. Campanero Martínez |
|---|---|
| Empresa | A3Z advanced S.L. |
| Cargo | Farmacéutica, Posgrado en Investigación en desarrollo, innovación de medicamentos (Universidad de Navarra). Becario de QA/RA - Doctor en Farmacia, Especialista en Farmacia Industrial y Galénica, Director Técnico, respect |
















Si continúas navegando, aceptas su uso.
Más información





Política de privacidad | Cookies | Aviso legal | Información adicional| miembros de CEDRO