














Aniol Gurrera
CEO, RAYPA

Almira Bartolomé
Directora de Business Development, 3PBIOVIAN

Jurgen Van Broeck
CEO, mAbxience



VARIACIONES: nuevas directrices

A partir del 15 de enero de 2026, entrarán en vigor las nuevas DIRECTRICES DE VARIACIONES DE LA COMISIÓN EUROPEA. Desde 2013, la Industria Farmacéutica ha tipificado las variaciones conforme a las directrices vigentes.
Sin embargo, en estos años, han sido muchas las novedades surgidas en la fabricación, procesos y recomendaciones aplicados sobre los medicamentos, además de nuevos productos como las terapias avanzadas (ATMPs), que han provocado la necesidad de una revisión global orientada a agilizar los trámites regulatorios.
Tanto la Industria Farmacéutica como la Agencia Europea del Medicamento (EMA) y las Agencias Reguladoras, celebran la publicación de estas nuevas directrices, cuya finalidad principal es optimizar la gestión del ciclo de vida de los medicamentos.
La reciente revisión de la Guía de Variaciones de la EMA (Reglamento de referencia 1234/2008) moderniza los procedimientos de cambio posautorización. Según la EMA, las nuevas directrices facilitarán un manejo más rápido y eficiente de las modificaciones, lo que beneficiará tanto a los Titulares de Autorizaciones de Comercialización (TAC) como a las Autoridades Reguladoras (AR).
Las variaciones abarcan cualquier modificación relacionada con una Autorización de Comercialización, incluyendo cambios administrativos, cambios en los procesos de fabricación, en la información del producto, indicaciones o actualizaciones de seguridad.
La revisión introduce una tramitación más rápida para los cambios de bajo riesgo y procedimientos más claros para la agrupación de solicitudes con el fin de mejorar la eficiencia y adaptarse al progreso científico. Los TAC deberán seguir presentando variaciones para cualquier modificación de la AC, con la aprobación de las AR cuando corresponda, para preservar la relación beneficio-riesgo.
En este ámbito, en el que la relación de las entidades públicas y privadas es constante, el objetivo final es incrementar la eficiencia y reducir la carga administrativa para la Industria Farmacéutica, optimizando a la vez los recursos de las autoridades competentes mediante procedimientos simplificados y optimizados, que garanticen los mismos niveles de calidad, eficacia y seguridad de los medicamentos.
Novedades Clave
Como se ha indicado, las nuevas directrices permitirán una tramitación más rápida. Entre los cambios relevantes destacan:
- Nuevas categorías de clasificación para problemas de seguridad inmediatos, Tipo 0, agilizando la aprobación de cambios de bajo riesgo.
- Agrupación anual voluntaria de las variaciones de Tipo IA.
- Modificación de los requisitos documentales y un nuevo énfasis en la agrupación de las variaciones.
- Nuevas herramientas como el Protocolo de Gestión de Cambios Posteriores a la Aprobación (PACMP), para rutas de cambio acordadas previamente y el Documento de Gestión del Ciclo de Vida del Producto (PLCM), para la supervisión continua de la autorización de comercialización.
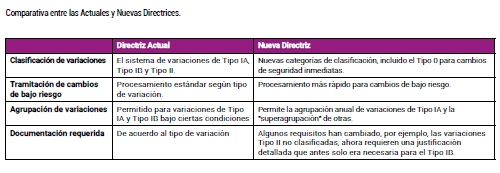
Clasificación de las variaciones:
Variaciones de Tipo IA (importancia menor, de bajo riesgo):
Igual que en las directrices actuales, el grupo de modificaciones tipo IA cubre cambios menores, como por ejemplo los administrativos. La novedad principal es la introducción de la agrupación anual voluntaria, que permite a los TAC agrupar varias variaciones de Tipo IA implementadas en un año natural en una sola notificación. Esta debe presentarse entre 9 y 12 meses después de la primera fecha de implementación. Se aplican excepciones para problemas urgentes de seguridad o actualizaciones de la información del producto.
Variaciones de Tipo IB (importancia menor, requieren aprobación):
Incluyen cambios como actualizaciones de seguridad acordadas o ajustes menores en la formulación. Los plazos de tramitación se acortan para agilizar las notificaciones, y se requieren justificaciones combinadas en los envíos agrupados.
Variaciones de Tipo II (importancia mayor):
Reservadas para cambios significativos (p. ej., nuevas indicaciones terapéuticas o modificaciones importantes en la fabricación), éstas mantienen los plazos de revisión estándar, pero se benefician de justificaciones simplificadas según el nuevo marco.
Variaciones de Tipo 0:
Se implementan para problemas de seguridad inmediatos e imprevistos (p. ej., retiradas urgentes de productos del mercado o mitigación de riesgos). Se implementan sin previo aviso, pero deben informarse retrospectivamente dentro de los plazos especificados, priorizando la respuesta rápida sobre una revisión exhaustiva.
Aspectos adicionales
Las nuevas directrices dan mayor importancia a las justificaciones sólidas, especialmente para las presentaciones agrupadas de Tipo IA. Para los cambios relacionados con la farmacovigilancia (por ejemplo, actualizaciones de QPPV), bastará con las notificaciones a través de la base de datos del Artículo 57, sin necesidad de presentar variaciones.
En los procedimientos nacionales, se permite la agrupación de variaciones de Tipo II y otras variaciones menores que afecten a múltiples autorizaciones de comercialización en el mismo Estado Miembro, siempre que sean iguales para todas las autorizaciones de comercialización y la autoridad nacional competente lo apruebe.
Los procedimientos de reparto de trabajo (worksharing) serán opcionales para las variaciones que incluyan al menos una variación de Tipo IB o Tipo II y afecten a múltiples autorizaciones de comercialización en diferentes Estados Miembros, incluso si involucran a diferentes TAC.
Para las variaciones implementadas antes del 15 de enero de 2026, seguirán aplicándose las directrices anteriores. La EMA publicará un documento de “Preguntas y respuestas” sobre el procedimiento y directrices técnicas, antes del 31 de diciembre de 2025, que abarcarán el uso de los eAF, las presentaciones combinadas y la gestión de variaciones imprevistas.
Implicaciones para el área de Regulatory Affairs
Esta actualización impacta directamente en la gestión regulatoria, aportando:
- Procedimientos de variación más rápidos, claros y ágiles.
- Clasificación basada en riesgo, con mayor precisión.
- Opciones de agrupación y compartición de trabajo más flexibles para variaciones en múltiples autorizaciones de comercialización.
- Uso estratégico del Protocolo de Gestión de Cambios post-aprobación (PACMP)
- Documento de Gestión del Ciclo de Vida del Producto (PLCM)
Las directrices ofrecen detalles sobre la aplicación de los procedimientos pertinentes, incluidlas medidas que deben adoptarse, desde la presentación de una notificación o una solicitud de modificación, hasta el resultado final del procedimiento.
Todo esto va de la mano con la era digital en la que vivimos. Los procedimientos regulatorios se digitalizan progresivamente y la Industria Farmacéutica tiene el deber de implementar prácticas de digitalización que estén a la vanguardia de la innovación en un entorno regulatorio en rápida evolución. Konexio Biotech está preparada para acompañar a las empresas en estos retos, ofreciendo soluciones estratégicas y soporte especializado para garantizar una transición eficiente y segura hacia este nuevo marco regulatorio.
Bibliografía:
- Reglamento (CE) nº 1234/2008 de la Comisión, de 24 de noviembre de 2008, relativo al examen de las modificaciones de los términos de las autorizaciones de comercialización de medicamentos para uso humano y medicamentos veterinarios.
- Directrices sobre los detalles de las diversas categorías de modificaciones, sobre el funcionamiento de los procedimientos establecidos en los capítulos II, II bis, III y IV del Reglamento (CE) n.o1234/2008 de la Comisión, relativo al examen de las modificaciones de los términos de las autorizaciones de comercialización de medicamentos para uso humano, y sobre la documentación que debe presentarse de conformidad con estos procedimientos (C/2025/5045).
| Nombre | Ángel Moreno Cela |
|---|---|
| Empresa | Konexio Biotech |
| Cargo | Regulatory Affairs |
















Si continúas navegando, aceptas su uso.
Más información





Política de privacidad | Cookies | Aviso legal | Información adicional| miembros de CEDRO